BIOL4585
Batch Processing & HPC Clusters
Overview
Last week, you performed association tests using plink, a .vcf file containing genotypes for 500 individuals, and the phenotypes for the 500 individuals. While this may seem like a lot of data, it only included the very small mitochondrial genome, which is ~17000 base pairs. The entire human genome is approximately two billion base pairs–over 100,000 times larger!
These kind of calculations will take more processing power than we’ve been using so far. If we tried to run a GWAS using a single processing core, it would take far too long. Luckily, Rivanna is a High-Performance Computing system made up of hundreds of processing cores that can be accessed by users. Today, we’ll learn how to access these powerful computing resources.
Specifically, you will learn:
- How to quickly view and modify text files using SFTP within a text editor. link
- Why it’s necessary to submit jobs to SLURM, instead of just running things interactively. link
- How core-hours represent the ‘currency’ of HPC systems. link
- How to submit and monitor jobs using the SLURM job manager. overview, writing your own
- How to submit dozens to hundreds of related tasks using job arrays. link
Reminder: don’t forget to update your course directory after logging in to Rivanna! Log in, cd into your directory on /scratch/ and then run
git fetch
git pull
Viewing and editing files with SFTP
Until now, you’ve viewed files using cat or less and edited files with vim or nano. While these commands are useful, sometimes all we want is to edit documents in a text editor. Now that you’ve learned how to do things the hard way, we’ll be learning a much simpler method, called SFTP. SFTP stands for SSH File Transfer Protocol, and that’s exactly what it does: transfers files over SSH. This will let you use a file browser within a text editor, so you can directly open, read, modify, and save text files in your Rivanna directory.
The text editor atom, developed by the team that made github.
-
Download and install the atom text editor
-
Open atom, and then open the settings menu under File/Settings or with they key combination
Ctrl+comma -
Select Install from the settings window and install the package ftp-remote-edit by the author h3imdall
-
Open the package with
Ctrl+spacebar -
You will be prompted to create a master password–this is an entirely new password, specific to the text editor. Think of it like a master password for accessing other servers.
-
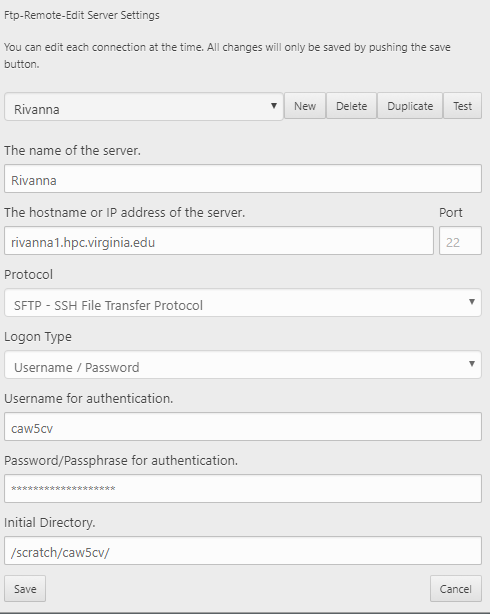
To access files on Rivanna, you need to enter information for connecting to it. Click edit servers to enter information. Yours should look similar to the settings below, but with your own computing ID and your own Rivanna password.
- Expand the
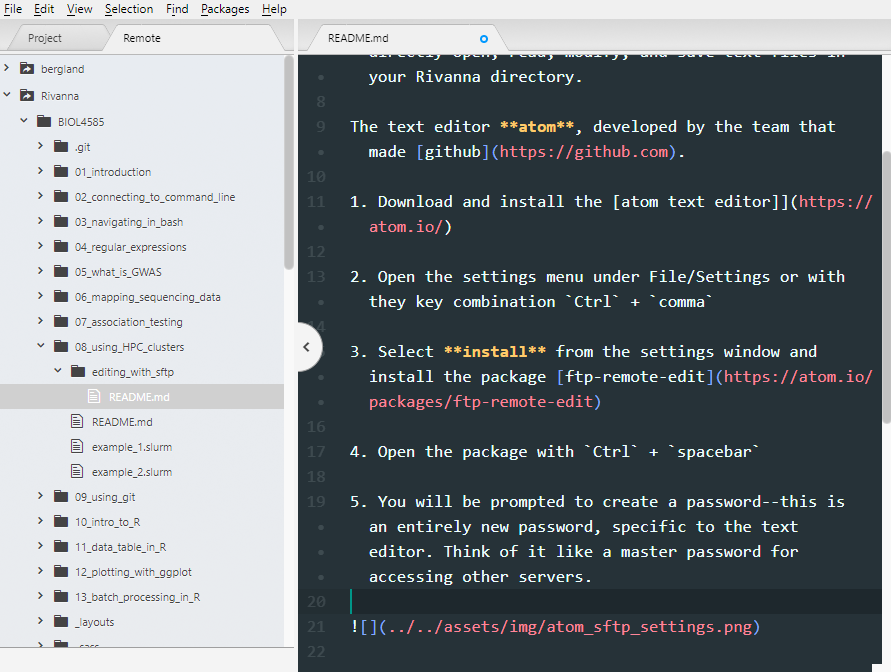
Rivannafolder to see the contents of your scratch directory within the file browser. You can open them to view or edit the contents. Upon saving, it will be uploaded to Rivanna. You can collapse and expand a directory to refresh its contents.
Job management on HPC systems
Many people use Rivanna at a single time. When running a program, it is typically necessary to reserve memory and processor cores. This is so the program doesn’t try to access resources that are currently in use or busy. when you log in, you have limited access to a set amount of memory and processor cores. You must submit a job to the SLURM job manager to access more resources. As an additional benefit, submitted jobs aren’t interrupted if your internet connection is closed. If you’re running a task interactively and your computer loses a connection, you’ll lose any progress and have to start over.
FYI, for those curious, SLURM stands for:
- Simple
- Linux
- Utility for
- Resource
- Management

No relation to Slurm MacKenzie of Futurama.
What are core-hours and service units?
Access to computing resources can be thought of as a service, and that costs resources. In order to make sure that access to Rivanna is shared across all users, instead of being hogged by few, access is ‘bought’ with a ‘currency’ called service units measured in core-hours: if you run a job on a single core for one hour, that job ‘costs’ one core-hour.
Typically, a research group will have an allocation of service units that everyone in the lab draws from. The allocation is like a shared banking account for service units. For this class, we all share an allocation that was granted to us for course use.
Overview of SLURM commands
SLURM takes user-provided information to best allocate computing resources. Jobs that request little memory and little time typically have a high priority. Jobs that request a significant amount of resources may need to ‘wait in line’ if resources are busy. All of these decisions of where and when jobs eventually run is handled behind the scenes. When you submit a job, you specify which queue it is ran in with --partition and one of the following options: standard, parallel, largemem or dev.
- the standard queue is the most common option, for typical use.
- the parallel queue is for jobs that require special architecture for parallel computing.
- the largemem queue is for jobs that need exceptionally large quantities of memory.
- the dev is for developing and testing scripts on a small scale. This queue doesn’t cost any core-hours when running, and is intended to be used to make sure code works, not for heavy use. After testing code in the dev queue, your tasks can be ran at the full scale on the other queues.
SLURM requests are formatted in bash scripts
The resource requests are usually included in the header of a bash script. View the contents of example.slurm to see how they are typically formatted. The parameters in these example files are:
--ntasksfollowed by an integer, the number of cores requested--memfollowed by the memory requested, e.g.8Gfor 8 gigabytes,120Mfor 120 megabytes--timefollowed by the time limit formatted asD-HH:MM:SS, e.g.0-12:00:00for 12 hours.--partitionfollowed by the queue, e.g.standardordev--accountfollowed by the allocation of service units to draw from
Note: the top line, formatted like #!/usr/bin/env bash is called a shebang line. It is used to identify what kind of language the file is written in, and ensures the file is executed using the right program. In this case, it’s written in bash and would be executed as such.
To submit a new job, use the sbatch command followed by the name of the slurm script to be submitted.
to view your recent job history, use the sacct command. Jobs typically have one of the following statuses:
| Job Status | Meaning |
|---|---|
| PENDING | Job has not started yet, and is waiting for resources |
| RUNNING | Job is currently running |
| FAILED | Job exited with errors |
| COMPLETED | Job exited without errors |
| CANCELLED | Job was cancelled by the user or an administrator |
| TIMEOUT | Job was terminated because it ran longer than requested |
To cancel a currently-running job, you can run scancel JOBID. JOBID is the number associated with a specific job, and you can view these IDs when you print your job history using sacct. You may wish to cancel a job if you realize there are mistakes in the code you submitted.
Writing and submitting SLURM scripts
- First, view the contents of the file
example.slurm. Notice there are missing values in the header. You will need to edit the values to request 1 core (--ntasks), 1G of memory (--mem), to run on the queuestandard(--partition), and to use the class’s allocation (--account, runallocationsto view which allocations you have access to). Then, submit the script as a job (shown below). The job should take a minute to run.
sbatch ./example.slurm
-
Check the progress of the job using
sacct, to see the progress change from pending to running to completed. -
View the messages printed during your job in the slurm output file. Any messages or errors that are printed during your job will be printed to a file named
slurm-JOBID.out. This means that you’ll need to manually view the contents of the file to see what was printed while the job ran. -
Submit the same script again. But this time, cancel the job using
scancelafter it starts. The JOBID is printed to your screen when you submit the job, and can also be seen in your job history (sacct). After cancelling,sacctwill show that the job status changed to CANCELLED.
Next, we’ll edit and test a script that maps sequencing reads, using the dev queue. The dev queue doesn’t cost any service units to run. However, jobs on the dev queue have a very low maximum time limit compared to others. You can see a list of available queues by running the command queues. We’ll use a smaller set of data for testing, to ensure that it runs quickly. Then, when we know it works, we can use the ‘full set’ of data. In this case, we’ll be mapping sequencing reads again, after downloading them with fastq-dump. Read through the map_reads.slurm
- Try submitting
map_reads.slurmwithsbatch. What happens? Why? - Make a copy of the
map_reads.slurmfile for editing (so we can keep the same original intact, just in case). - Insert an appropriate SLURM header to this newly-created file. You’ll only need a single core (
--ntasks), 8G of memory, and 45 minutes of time. You’ll want to use thedevqueue - Submit this newly edited script. Check job progress with
sacct. - Diagnose and fix any problems that occur–there may be an ‘accidental’ bug in the code. The
slurm-<JOBID>.outfile may give hints of what is happening. Resubmit after you think you’ve fixed the problem; continue until the job runs to completion.
Still not working? Double-check that you are working in the directory /scratch/$USER/BIOL4585/08_using_HPC_clusters. It seems to be a somewhat common error that the BIOL4585 repository was downloaded twice, resulting in something like /scratch/$USER/BIOL4585/BIOL4585/08_using_HPC_clusters (notice BIOL4585 exists twice). If this is the case for you, you can rectify the situation as follows:
# first, rename the 'outer' folder
mv /scratch/$USER/BIOL4585/ /scratch/$USER/BIOL4585_old/
# second, move the 'inner' folder up one level
mv /scratch/$USER/BIOL4585_old/BIOL4585/ /scratch/$USER/
# From here, you can either leave the old folder alone, or remove it via
rm -rf /scratch/$USER/BIOL4585_old/
Scaling up to the full-sized job
Congratulations! The testing phase with the subset o
Create another copy of map_reads.slurm as a new file for the full-sized run. You can then edit this file in the following ways:
- Remove
-X <integer>from thefastq-dumpline so the full file is downloaded, instead of just the firstsequencing reads - Create a SLURM header that requests at leats 1 hour of time, 4 cores (tasks), and 32G of memory on the
standardqueue (instead ofdev) - Change the bwa-mem line to use 4 cores (
-toption followed by # of cores) - HaplotypeCaller will automatically use all available cores
- Change the value of the
subfoldervariable to something to identify this is the real, full run including all data - Submit the job to SLURM. You can view the job status with
sacctand view the printed output as it’s running vialess slurm-<jobid>.out - While waiting for the job to run (it might take 20+ minutes), it may be a good time to work through in-class questions.
- A fully successfull run should produce no errors, and an output
MT.vcffile.
Using job arrays
Sometimes you might want to submit a large number of similar jobs. For example, if we wanted to run the map_reads.slurm script for hundreds of different .fastq files. Instead of having to run sbatch over and over again, you can submit a job array. If you wanted to submit 700 iterations of a script filename.slurm, but run no more than 10 at a time, you could run:
sbatch --array=1-700%10 filename.slurm
Try running a job array yourself: Edit the array_job.slurm file to run with a single core, 1G of memory, a 5 minute time request, on the standard queue. Then, submit array_jobs.slurm as an array of 50 jobs, limited to 5 at a time. Once submitted, use sacct to track the progress of your jobs.
In-class questions (answer on collab before leaving class):
- What’s the difference between the standard and dev queues–when would you use each?
- How many core-hours would it cost to run a single job with 4 cores for 45 minutes?
- Run the command
allocations. Which allocation(s) you have permissions to use when submitting jobs, and how many service-hours does it have? - You view the contents a file and notice the first line is
#!/usr/bin/env python. What is this kind of line called, and what does it tell you about the file? What does this tell the shell about the file? - How does editing files in atom using sftp compare to editing files using
vimornano? Which do you prefer and why? - You check your job progress using
sacctand see that one of your job’s status is FAILED. Where would you first look to diagnose the problem? - Examine the below lines, from the
map_reads.slurmfile. Until now you haven’t seen&&or||used before. What do these symbols accomplish and why might it be useful?
mkdir -p /scratch/$USER/BIOL4585/08_using_HPC_clusters/${subfolder} && cd /scratch/$USER/BIOL4585/08_using_HPC_clusters/${subfolder}
cp ../MT_reference.FASTA . || { echo "could not copy MT_reference.fasta to working directory"; exit 1; }
Homework
Complete the Week 08 questions on the Tests & Quizzes tab of Collab.
The early submission deadline is Friday Night at 11:59 PM. Final submission deadline is the start of class next week.
Feedback will be available Saturday by no later than noon. Any questions that were wrong can be submitted to me via email for up to half credit back on incorrect answers, if submitted prior to the start of next class.